Academics
| Degree | University/Institution |
|---|---|
| Ph.D. in Science (Microbiology) | Institute: National Institute of Cholera and Enteric Diseases (NICED), Kolkata. Guide: Dr. G. Balakrish Nair University: Jadavpur University, Kolkata. Co-Guide: Prof. Jasmina Khanam |
Work Experience
| Position | University/Organisation | Period |
|---|---|---|
| Research Associate | Loyola University Medical Center | Jan 2000 to April 2001 |
| Research Associate | University of Illinois at Chicago | May 2001 to June 2002 |
| Instructor | University of Illinois at Chicago | June 2002 to June 2003 |
| Research Assistant Professor | University of Illinois at Chicago | June 2003 to Jan 2005 |
| Lecturer/Scientist-B | Institute of Life Sciences | Feb, 2005 to May, 2007 |
| Scientist-C | Institute of Life Sciences | June, 2007 to May, 2011 |
| Scientist-D | Institute of Life Sciences | June, 2011 to June, 2015 |
| Scientist-E | Institute of Life Sciences | July, 2015 to June, 2020 |
| Scientist-F | Institute of Life Sciences | July, 2020 to September, 2025 |
| Scientist-G | Institute of Life Sciences | October, 2025 to date |
Awards & Recognition
| Details |
|---|
ICMR International Fellowship, Feb to July 2011 |
Research
| Details | |||||||
|---|---|---|---|---|---|---|---|
Two broad themes drive the research activities of the lab,
A complete understanding of the role of specific regulators (known or novel) in the progression of a disease can profoundly impact the diagnosis, therapy, and, ultimately, the survival of the patients. In this regard, we look into the molecular mechanisms that delineate CML stem cells from normal hematopoietic stem cells. The research findings may lead to the development of targeted molecular therapies to eliminate the disease before it progresses.
Post-translational protein modification is essential in multiple cellular processes, including DNA repair, protein stability, nuclear translocation, protein-protein interactions, cellular proliferation, differentiation, and apoptosis. Multiple post-translational modifications on a protein constitute a complex regulatory program that transduces molecular information to and from signaling pathways. We are investigating whether post-translational modification(s) of EVI1 promote leukemogenesis/oncogenesis. Projects are available to study the
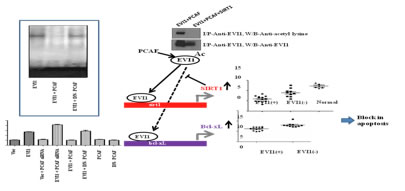
The lab is always open to any new ideas by a student, a post-doc, or a researcher, as long as they fall within the themes that drive the lab’s research activities. Our contributions Acetylation of the proto-oncogene EVI1 abrogates Bcl-xL promoter binding and induces apoptosis Pradhan et al., 2011 In this study, we provide evidence that EVI1 directly induces the expression of Bcl-xL through the first set of zinc finger, thereby inhibiting apoptosis. ChIP analysis showed that EVI1 binds to the Bcl-xL promoter in HT-29 cells, a colon carcinoma cell line expressing EVI1. The observation is also supported by the fact that EVI1 siRNA-treated HT 29 cells show a downregulation of Bcl-xL expression and that over-expression of EVI1 results in the induction of the Bcl-xL reporter construct. A set of EVI1 positive chronic myeloid leukemia (CML) samples also showed higher Bcl-xL expression with respect to EVI1 negative samples. Interestingly, co-expression of EVI1 with wild type, but not with dominant-negative form of PCAF, abolishes the effect of EVI1 on Bcl-xL, indicating that acetylation of EVI1 abrogates its ability not only to bind Bcl-xL promoter but also alleviate Bcl-xL activity. Finally we have shown that EVI1 expression regulates apoptosis in HT-29 cells, which is abrogated when HT-29 cells are transfected with EVI1 siRNA or PCAF. The result for the first time shows a direct pathway by which EVI1 can protect cells from apoptosis and also demonstrates that the pathway can be reversed when EVI1 is acetylated. EVI1 up-regulates the stress responsive gene SIRT1 which triggers deacetylation and degradation of EVI1 Pradhan et al., 2011 In this report, we show that SIRT1, a histone deacetylase is a direct target of EVI1. In vivo chromatin immunoprecipitation assay revealed that EVI1 binds to the promoter region of SIRT1 approximately 1 kb upstream of the transcription start site. The functionality of the site was deduced by luciferase assay which showed that EVI1 significantly increases the SIRT1 promoter activity. SIRT1 was also found to be up regulated in cell lines and in chronic myeloid leukemia patient samples where EVI1 was detected. Over expression of SIRT1 in cells shows that it interacts with EVI1 and this interaction lead to the deacetylation of the protein. Upon deacetylation the stability of EVI1 was found to be affected which was negatively regulated by nicotinamide (NAM). Our results thus identify an EVI1-SIRT1 axis in the regulation of EVI1 activity suggesting a possible role of SIRT1 in EVI1 positive neoplasms.
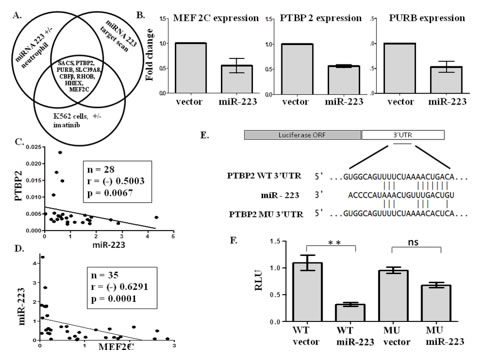
BCR-ABL mediated repression of miR-223 results in the activation of MEF2C and PTBP2 in chronic myeloid leukemia Agatheeswaran et al., 2012 We have reported here that miR-223 downregulation affects the transcription factor MEF2C and alternative splicing factor PTBP2. Our results suggest that changes in the miR-223/PTBP2 pathway can contribute to an abnormal splicing of several genes and shed light into the potential role exerted by miRNAs in a subset of CML. Not only did it highlight the ability of miRNAs to alter mRNA but also, more importantly, it added a new layer to the complexity of mechanisms regulating the phenotype of CML.
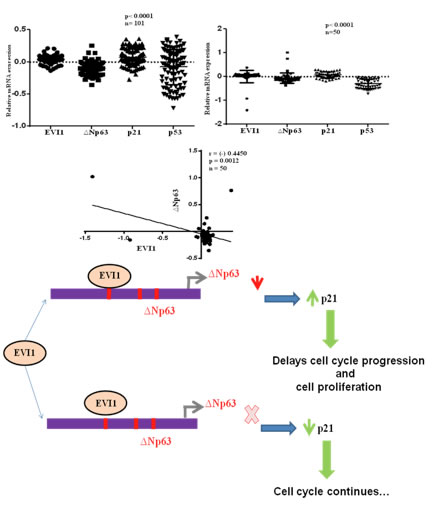
EVI1 targets ∆Np63 and upregulates the cyclin dependent kinase inhibitor p21 independent of p53 to delay cell cycle progression and cell proliferation in colon cancer cells Nayak et al., 2013 Our data for the first time shows that ecotropic viral integration site I binds to ∆Np63 promoter element directly and down regulates its expression. Down regulation of ∆Np63 induces the expression of p21 in HT-29cells and also in colon carcinoma cells that do not express p53 including patient samples expressing low level of p53, that eventually delay cell cycle progression at G0/G1 phase. Concomitant silencing of ecotropic viral integration site I from the cells or introduction of ∆Np63 to the cells significantly rescued this phenotype, indicating the growth defect induced by ∆Np63 deficiency to be, at least in part, attributable to ecotropic viral integration site I function. The mutual regulation between ecotropic viral integration site I and ∆Np63 may constitute a novel axis which might affect the downstream pathways in cells that do not express functional p53.
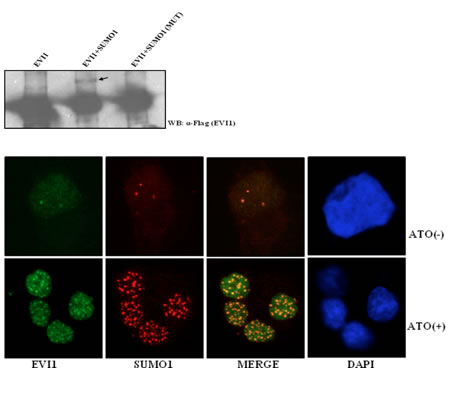
SUMO1 negatively regulates the transcriptional activity of EVI1 and significantly increases its co-localization with EVI1 after treatment with arsenic trioxide Singh et al., 2013 Aberrant expression of the proto-oncogene EVI1 (ecotropic virus integration site1) has been implicated not only in myeloid or lymphoid malignancies but also in colon, ovarian and breast cancers. Despite its importance in oncogenesis, the regulatory factors and mechanisms that potentiate the function of EVI1 and its consequences are partially known. Here we demonstrated that EVI1 is post-translationally modified by SUMO1 at lysine residues 533, 698 and 874. Although both EVI1 and SUMO1 were found to co-localize in nuclear speckles, the sumoylation mutant of EVI1 failed to co-localize with SUMO1. Sumoylation abrogated the DNA binding efficiency of EVI1 and also affected EVI1 mediated transactivation. The SUMO ligase PIASy was found to play a bi-directional role on EVI1, PIASy enhanced EVI1 sumoylation and augmented sumoylated EVI1 mediated repression. PIASy was also found to interact with EVI1 and impaired EVI1 transcriptional activity independent of its ligase activity. Arsenic trioxide (ATO) known to act as an anti leukemic agent for acute promyelocytic leukemia (APL) not only enhanced EVI1 sumoylation but also enhanced the co-localization of EVI1 and SUMO1 in nuclear bodies distinct from PML nuclear bodies. ATO treatment also affected the Bcl-xL protein expression in EVI1 positive cell line. Thus, the results showed that arsenic treatment enhanced EVI1 sumoylation, deregulated Bcl-xL, which eventually may induce apoptosis in EVI1 positive cancer cells. The study for the first time explores and reports sumoylation of EVI1, which plays an essential role in regulating its function.
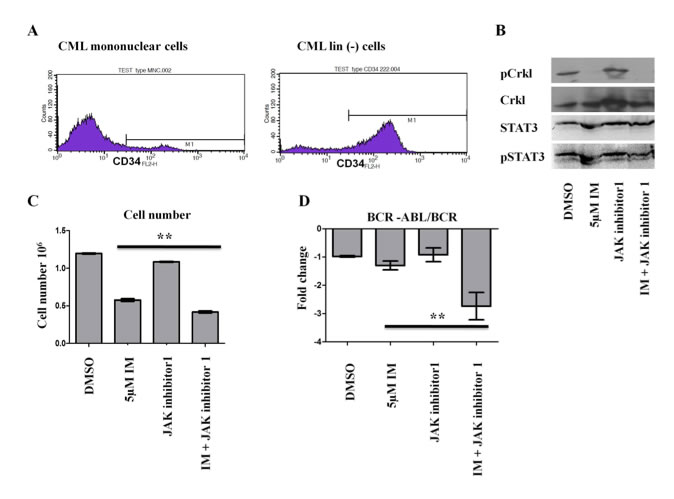
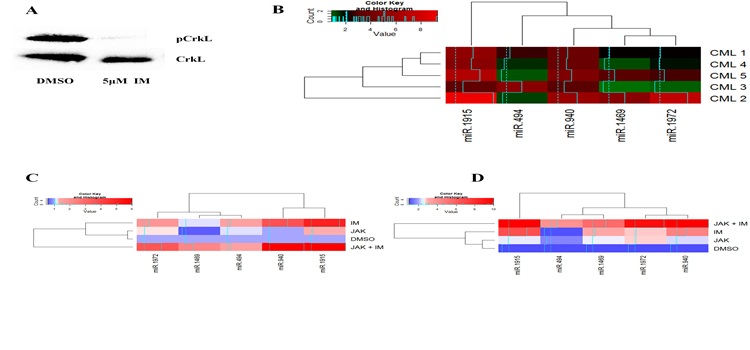
JAK inhibitors along with TKIs can eradicate CML lineage negative cells Agatheeswaran et al., 2014 CML lin(-) cells from the bone marrow of naive CML cases were purified by using CML debulking kit. Approximately 80% of the lin(-) cells were found to be positive for CD34 marker. We observed that imatinib efficiently blocks the BCR-ABL kinase activity but failed to eliminate the CML lin(-) cells in an in vitro culture system when supplemented with cytokines. However, a combination of imatinib and JAK inhibitor 1 brought down the cell proliferation significantly. The combination of the imatinib and JAK inhibitor 1 also showed a significant decrease in BCR-ABL/BCR ratio with respect to imatinib alone. Our results suggests the fact that combination of TKI inhibitor along with a JAK inhibitor can efficiently eliminate CD34+ CML cells and in the process residual normal stem and progenitor cells can expand considerably.
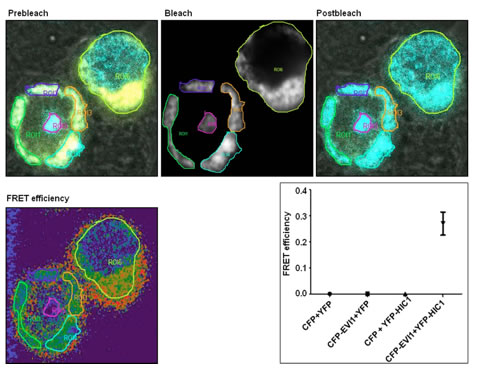
Physical and functional interaction of the proto-oncogene EVI1 and tumor suppressor gene HIC1 deregulates Bcl-xL mediated block in apoptosis Pradhan et al., 2014 Several studies have established ecotropic viral integration site 1 as both a transcription factor and an interacting partner that presumably regulates gene expression. Using coimmunoprecipitation and fluorescence resonance energy transfer analysis, we found that the N-terminal domain of hypermethylated in cancer 1 interacts with the proximal set of zinc fingers of ecotropic viral integration site 1.This interaction not only abolishes the DNA binding activity of ecotropic viral integration site 1 but also disrupts the transcriptional activity of an anti-apoptotic gene promoter selectively targeted by ecotropic viral integration site 1. By using flow cytometry and western blotting, here we show that hypermethylated in cancer 1 can deregulate ecotropic viral integration site 1 mediated blockage of apoptosis. We hypothesize that therapeutic upregulation of hypermethylated in cancer 1 may provide an important means of targeting ecotropic viral integration site 1 positive cancers.
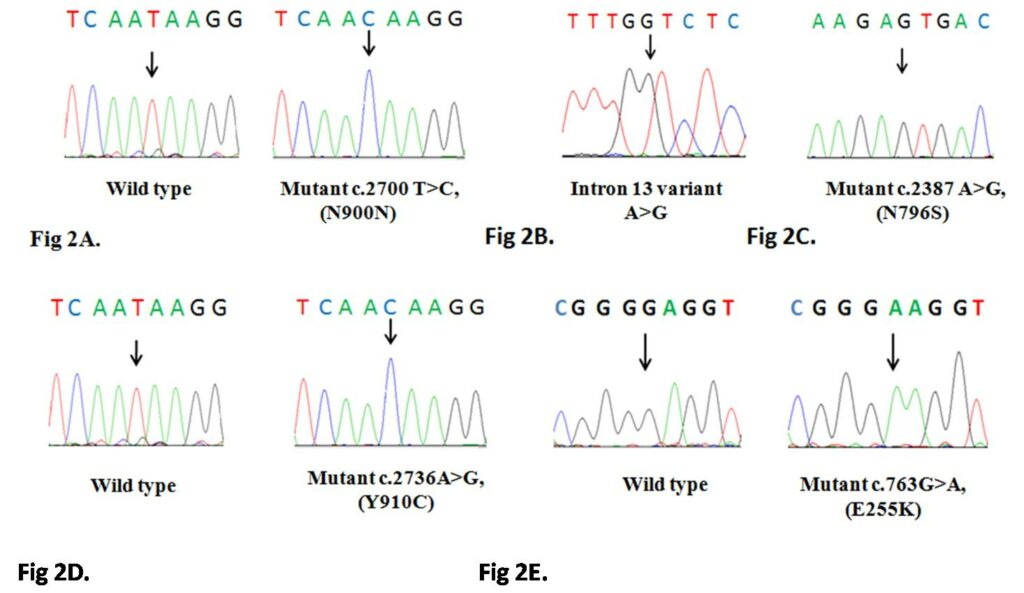
Dual transcripts of BCR–ABL and different polymorphisms in Chronic Myeloid Leukemia patients from Odisha, India Nandagopalan et al., 2015 Chronic myeloid leukemia is characterized by the presence of a hallmark chromosomal translocation, the Philadelphia chromosome. It is the commonest form of adult leukemia in the Indian population. Although there is no dearth of reports regarding the different variants of BCR-ABL in the literature, we studied the co-expression of e13a2 and e14a2 transcripts and few polymorphisms in CML patients from Odisha, India. Molecular genetics approach was adapted to screen for polymorphisms, mutation and translocation in BCR, ABL kinase domain and BCR-ABL breakpoint region in 73 CML patients. All 8 patients with dual transcripts were found to harbor an exonic polymorphism (c.2700 T>C) and an intronic polymorphism (g.109366A>G) that were earlier reported to be associated with co-expression of both the transcripts. We also observed c.763G>A mutation in ABL kinase domain that confers reduced sensitivity to tyrosine kinase inhibitors and two polymorphisms, c.2387 A>G and c.2736A>G in the BCR gene. Although our data supports the previous findings that co-expression of BCR-ABL transcripts is due to the occurrence of exonic and intronic polymorphisms in the BCR gene it also shows that the intronic polymorphism can arise without the linked exonic polymorphism in the Indian population. The occurrence of ABL kinase domain mutation is less frequent in Indian population. We also report the presence of N796S polymorphism that was earlier reported to be associated with bipolar disorder; however we were not able to find any significant correlation between CML disease occurrence and the polymorphism, its frequency was similar in the control population.
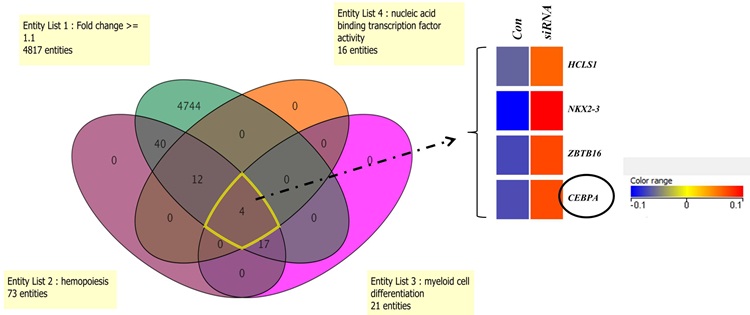
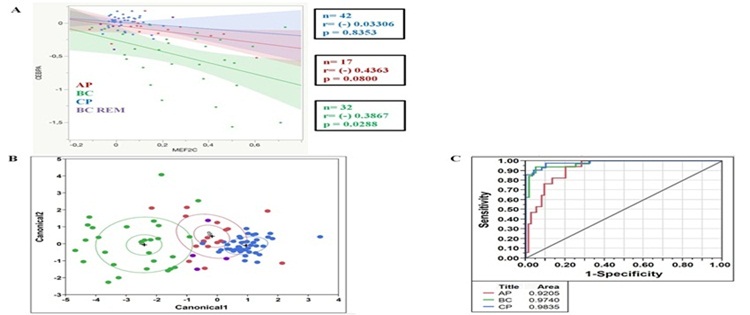
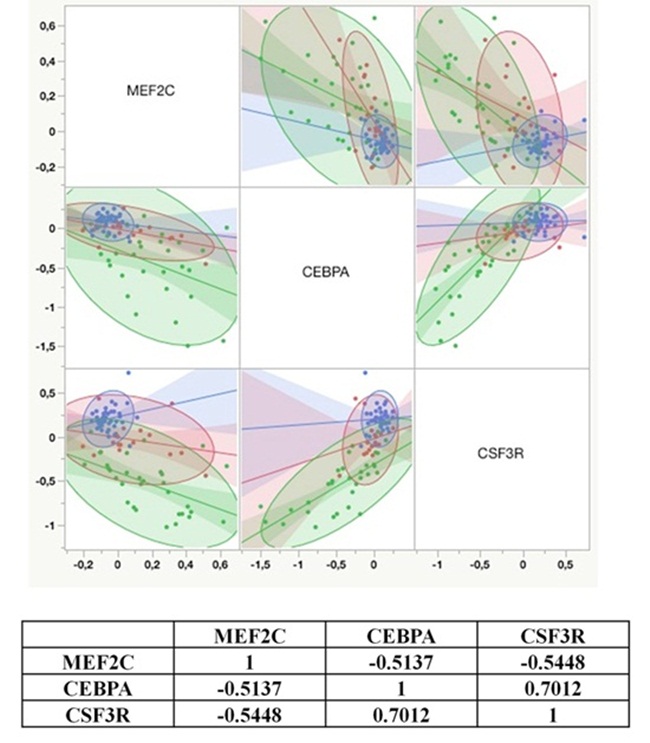
MEF2C and CEBPA: possible co-regulators in Chronic Myeloid Leukemia disease progression Agatheeswaran S and Chakraborty S, 2016 Chronic myelogenous leukemia (CML), a hematopoietic malignancy, characterized initially by a chronic phase (CP) progresses into blast crisis (BC) with the accumulation of secondary abnormalities. We have reported earlier that MEF2C, a target of miR-223, was significantly up regulated in CML and also showed a negative correlation with miR-223. In this study, gene expression arrays were used to identify the genes regulated by MEF2C during myelopoiesis. Statistical tools were used to understand the correlation between MEF2C and the targets in different phases of CML. Different CML cell lines and CML patient samples were treated with imatinib to study the effect of MEF2C on the target genes. We observed that MEF2C targets a set of myeloid genes including the myeloid transcription factor CEBPA. MEF2C and CEBPA expression patterns are negatively correlated in CML patient samples. We further show that the expression of MEF2C and CEBPA along with CSF3R is sufficient to molecularly classify different stages of CML. Imatinib, the drug of choice for CML, abrogates MEF2C expression and reverses CEBPA repression both in the cell line and the primary cells. We report the existence of a MEF2C and CEBPA correlation in CML disease progression.
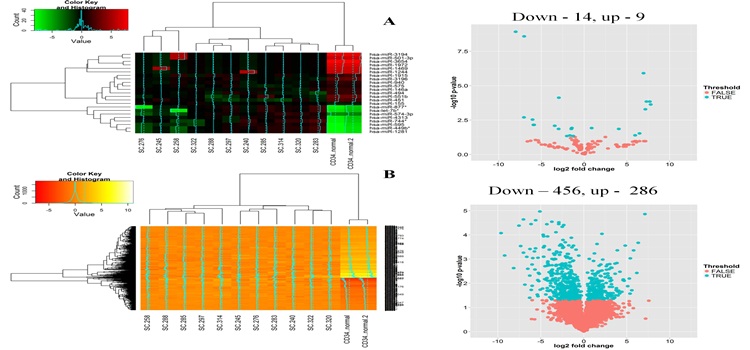
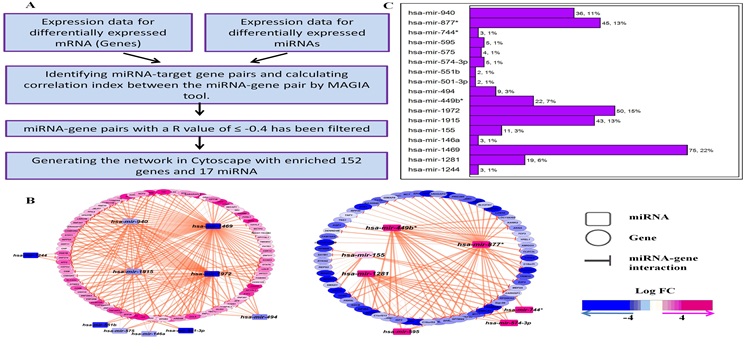
Identification and functional characterization of the miRNA-gene regulatory network in chronic myeloid leukemia lineage negative cells Agatheeswaran S, 2016 Chronic myeloid leukemia (CML) is maintained by leukemic stem cells (LSCs) which are resistant to the existing TKI therapy. Hence a better understanding of the CML LSCs is necessary to eradicate these cells and achieve complete cure. Using the miRNA-gene interaction networks from the CML lin(-) cells we identified a set of up/down-regulated miRNAs and corresponding target genes. Association studies (Pearson correlation) from the miRNA and gene expression data showed that miR-1469 and miR-1972 have significantly higher number of target genes, 75 and 50 respectively. We observed that miR-1972 induces G2-M cell cycle arrest and miR-1469 moderately arrested G1 cell cycle when overexpressed in KCL22 cells. We have earlier shown that a combination of imatinib and JAK inhibitor I can significantly bring down the proliferation of CML lineage negative cells. Here we observed that Imatinib and JAK inhibitor I combination restored the expression pattern of the down-regulated miRNAs in primary CML lin(-) cells. Thus effective manipulation of the deregulated miRNAs can restore the miRNA-mRNA networks that can efficiently inhibit CML stem and progenitor cells and alleviate the disease.
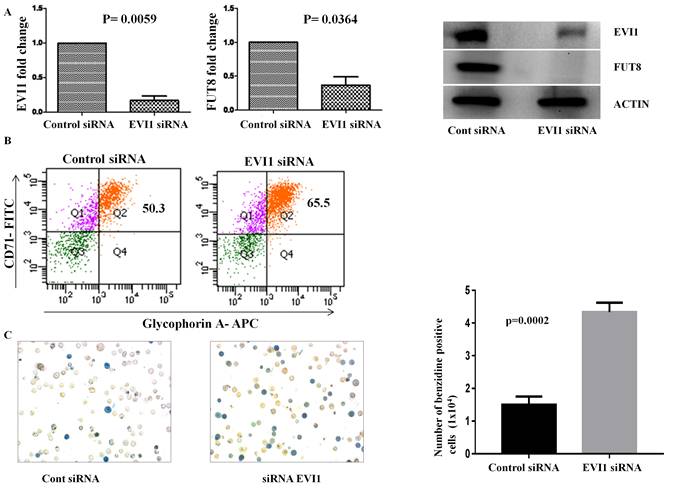
Ecotropic viral integration site I regulate alpha-1, 6 fucosyl transferase expression and blocks erythropoiesis in chronic myeloid leukemia Kuila et al., 2016 Although BCR-ABL is the hallmark genetic abnormality of chronic myeloid leukemia (CML), secondary molecular events responsible for the evolution of the disease to blast crisis are yet to be deciphered. Taking into account the significant association of EVI1 in CML drug resistance it is necessary to decipher the other roles played by EVI1 in CML disease progression. In this regard we cross-hybridized three microarray datasets and deduced a set of 11 genes that seems to be regulated by EVI1 in CML. We observed a strong correlation between EVI1 and FUT8 in the chronic phase of the disease and both of them were found to be up regulated with the progression of the disease. Knockdown of EVI1 in a CML cell line not only down regulated FUT8, but also rendered the cells towards erythroid differentiation. Our study shows the involvement of EVI1 and FUT8 axis in blocking erythropoiesis in CML.
Ecotropic viral integration site 1 promotes metastasis independent of epithelial mesenchymal transition in colon cancer cells Nayak et al., 2018 The most indecipherable component of solid cancer is the development of metastasis which accounts for more than 90% of cancer-related mortalities. A developmental program termed epithelial-mesenchymal transition (EMT) has also been shown to play a critical role in promoting metastasis in epithelium-derived solid tumors. By analyzing publicly available microarray datasets, we observed that ecotropic viral integration site 1 (EVI1) correlates negatively with SLUG, a master regulator of EMT. This correlation was found to be relevant as we demonstrated that EVI1 binds to SLUG promoter element directly through the distal set of zinc fingers and downregulates its expression. Many studies have shown that the primary role of SLUG during EMT and EMT-like processes is the regulation of cell motility in most of the cancer cells. Knockdown of EVI1 in metastatic colon cancer cell and subsequent passage through matrigel not only increased the invading capacity but also induced an EMT-like morphological feature of the cells, such as spindle-shaped appearance and led to a significant reduction in the expression of the epithelial marker, E-CADHERIN and increase in the expression of the mesenchymal marker, N-CADHERIN. The cells, when injected into immunocompromised mice, failed to show any metastatic foci in distant organs however the ones with EVI1, metastasized in the intraperitoneal layer and also showed multiple micro metastatic foci in the lungs and spleen. These findings suggest that in colon cancer EVI1 is dispensable for epithelial-mesenchymal transition, however, is required for metastasis. 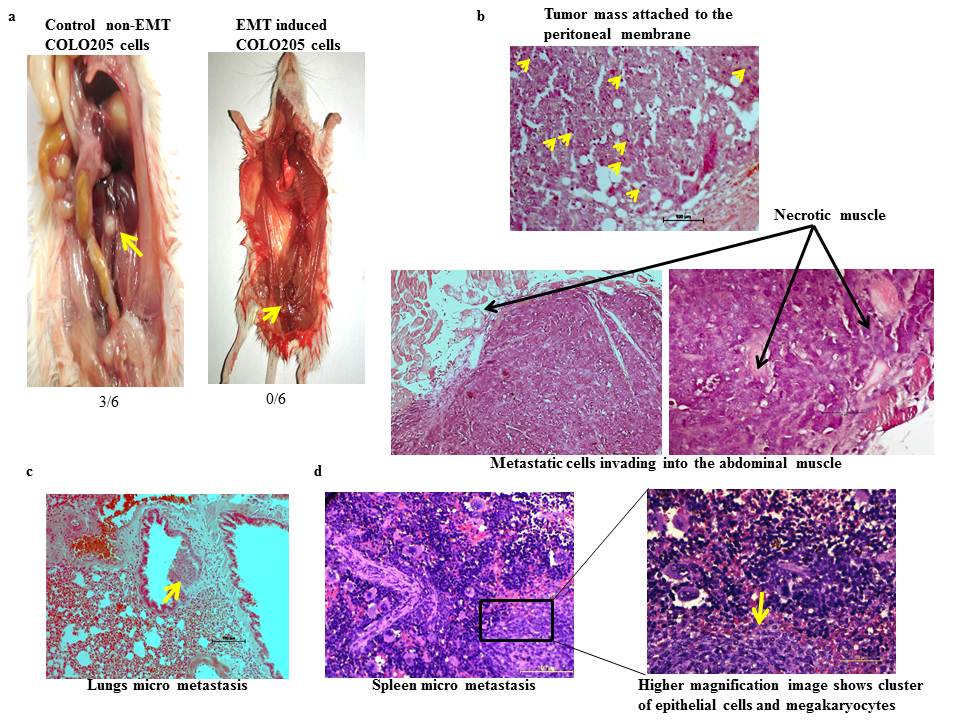 PTBP2 exon 10 inclusion is associated with the progression of CML and it is BCR-ABL1 dependent
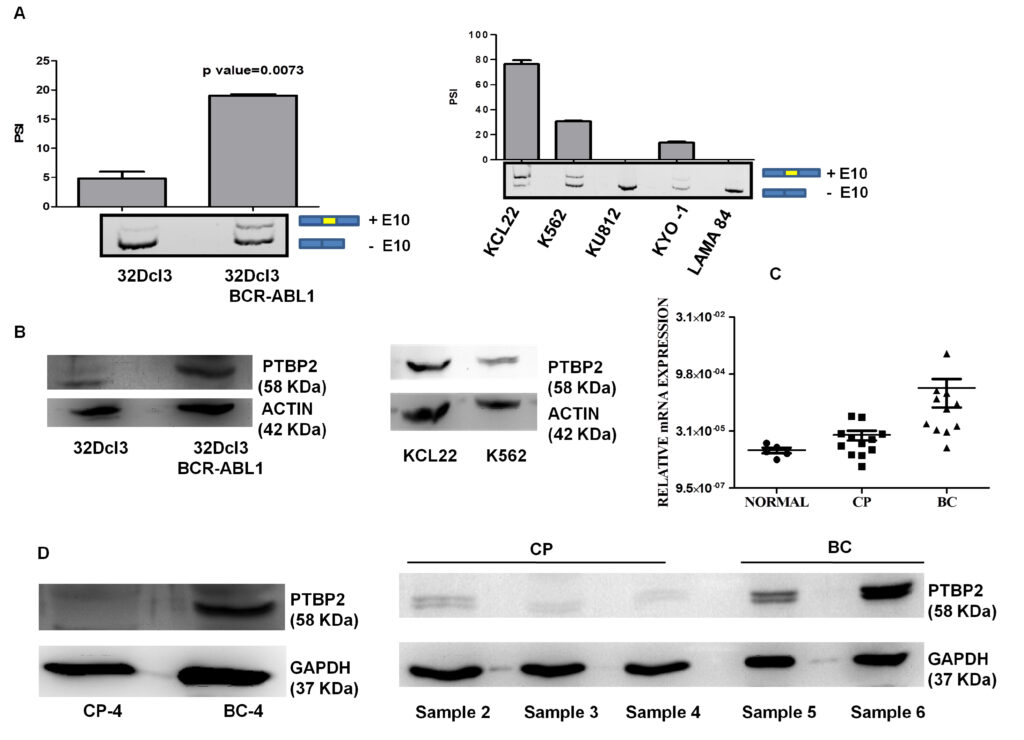
Nandagopalan et al., 2019 Altered or aberrant expression of several splicing factors leads to the progression of different cancers. Though there are several ongoing studies underscoring the role of the splicing regulator polypyrimidine tract binding protein 2 (PTBP2) in neuronal cells, we unveil the role of PTBP2 in chronic myeloid leukemia (CML). Different RNA binding proteins (RBP’s) earlier reported in chronic myeloid leukemia blast crisis (CML-BC) cases (n=28) from Radich oncomine leukemia dataset, were compared. We observed increased expression of MSI2 followed by PTBP2 in BC cases and increased PTBP2 expression in relapsed cases (n=10) from the same dataset compared to other RBP’s. We also observed increased PTBP2 exon 10 inclusion in KCL22, a granulocytic lineage CML cell line when compared to other CML cell lines of different lineages. As PTBP2 protein expression is associated with PTBP2 exon 10 inclusion, we observed in cell lines and in a set of progressed cases (n=4) that increased BCR-ABL1 expression potentiates PTBP2 exon 10 inclusion and thus confers the existence of a functional protein. Inhibition of BCR-ABL1 with imatinib not only blocks the inclusion of exon 10 but also deregulates PTBP2 expression in CML cells. Knockdown of PTBP2 in KCL22 cells leads to reduced cell proliferation, increased G2/M cell cycle arrest and increased apoptosis. Taken together our study portrays PTBP2 as a new possible target for CML and progressive inclusion/exclusion of PTBP2 exon 10 in CML might play an important role CML progression.
EVI1 Promotes Metastasis by Downregulating TIMP2 in Metastatic Colon and Breast Cancer Cells
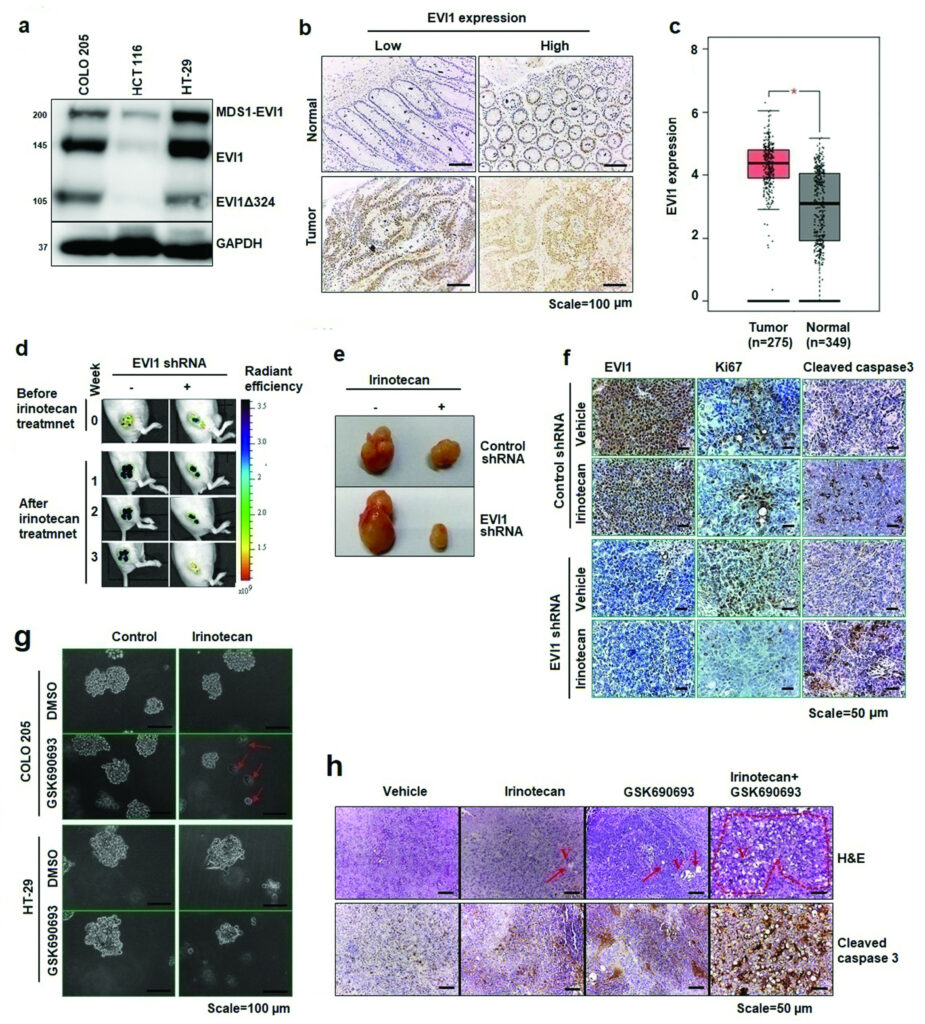
Pradeepa et al., 2022 Ecotropic viral integration site-1 (EVI1) is an oncogenic zinc finger transcription factor whose expression is frequently upregulated in various cancers, including myeloid malignancies and solid tumors. Previously, our group has shown that EVI1 knockdown minimizes colon cancer cells’ metastatic potential compared to control cells. In this study, control and EVI1 knockdown colon cancer cells were subjected to the microarray to identify the potential targets that regulate cancer metastasis. Differential gene expression analysis revealed significant downregulation of tissue inhibitor of matrix metalloproteinase-2 (TIMP2) in EVI1 expressing cells. EVI1 knockdown increased TIMP2 protein expression levels and reduced wound healing and migration capacity in metastatic cells. Mechanistically, the TIMP2 promoter harbors potential binding sites for EVI1; EVI1 binds to the TIMP2 promoter and represses its expression, as observed using ChIP and luciferase assay. TIMP2 is an important metastasis suppressor gene; however, its function is suppressed in many cancers through hypermethylation. Thus, demethylation could prove to be a potential alternative to reactivate TIMP2 functional activity. Immunoprecipitation analysis showed that DNA-methyltransferase 1 (DNMT1), which plays a vital role in maintaining the genome methylation pattern during DNA replication and repair, interacts with EVI1 to promote TIMP2 silencing. Treating cancer cells in vitro with a known demethylation agent, 5-aza-2′-deoxycytidine (Aza-D), restored the optimal TIMP2 expression without altering EVI1 binding efficiency and reduced relative wound healing potential of cancer cells. Animal studies showed that Aza-D treated cells injected through the intravenous route exhibited reduced liver and skin metastasis compared to non-treated cells. Furthermore, Aza-D treatment in mice delayed the metastasis progression compared to the vehicle-treated group. Thus, the present study provides an insight into the therapeutic applications of demethylating agents to reduce cancer metastasis in models with EVI1 overexpressing tumors. AKT inhibition sensitizes EVI1 expressing colon cancer cells to irinotecan therapy by regulating the AKT/mTOR axis
Pradeepa et al. 2022 The functional role of Ecotropic Viral Integration site 1 (EVI1) in colon cancer drug resistance is not well understood. In the present study, the role of EVI1 in irinotecan resistance was evaluated using different colon cancer models. Knocking down of EVI1 decreased cancer stem cell-like properties and improved the irinotecan response in both cell line and nude mice subcutaneous xenograft model. Further investigation showed that downregulation of EVI1 resulted in the inhibition of AKT/mTOR signaling. In addition, EVI1 knockdown decreased overall expression of the rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR), one of the significant components of mTORC2 complex required for full activation of AKT. Knocking down RICTOR protein expression further increased the irinotecan cytotoxic effects in EVI1 downregulated cancer cells. Treating colon cancer cells with ATP-competitive AKT inhibitor GSK690693 increased the therapeutic potential of irinotecan by altering AKT kinase activity. Co-treatment with irinotecan and GSK690693 reduced 3D structure formation and cell viability, increased apoptosis in cell line models, and decreased relative tumor progression rate and size in animal models. Further, co-treatment of irinotecan with GSK690693 decreased phospho-mTOR, phospho-GSK-3β, RICTOR level, and increased cleaved caspase 3, a marker of apoptosis. These results demonstrated the potential role of the EVI1-AKT/mTOR axis in irinotecan resistance. In addition, inhibition of the AKT signaling cascade by using GSK690693 could be a possible alternative to improve the irinotecan response in EVI1-expressing colon cancer models.
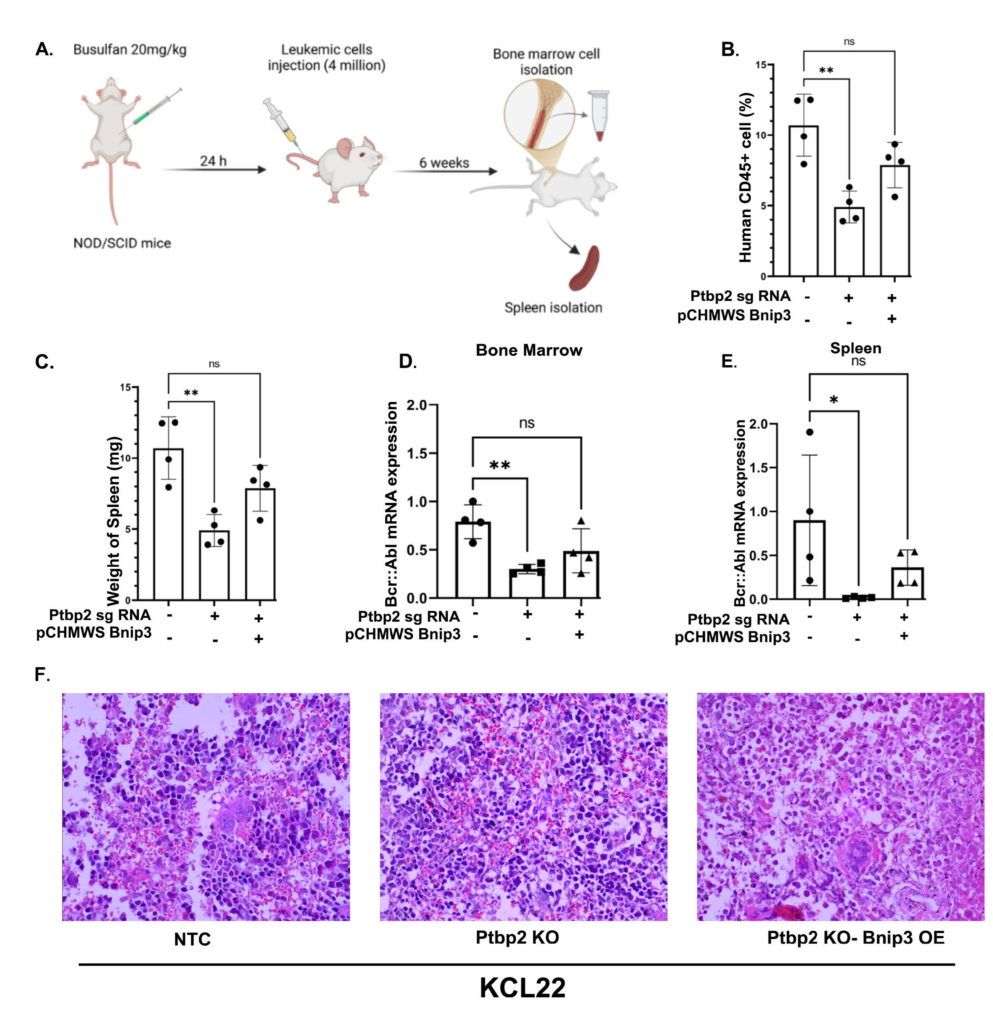
PTBP2 promotes cell survival and autophagy in Chronic Myeloid Leukemia by stabilizing BNIP3 Barik et al., 2025 Polypyrimidine tract binding protein 2 (PTBP2) regulates alternative splicing in neuronal, muscle, and Sertoli cells. PTBP2 and its paralog, PTBP1, which plays a role in B-cell development, were found to be expressed aberrantly in myeloid leukemia. Genetic ablation of Ptbp2 in the cells resulted in decreased cellular proliferation and repopulating ability, decreased reactive oxygen species (ROS), and altered mitochondrial morphology. RNA immunoprecipitation followed by sequencing (RIP-seq) and functional assays confirmed that PTBP2 binds to Bcl-2 Interacting Protein 3 (Bnip3)-3’UTR and stabilizes its expression.
Our study also suggests that PTBP2 promotes autophagy, as evidenced by the low levels of LC3-II expression in Ptbp2-knockout cells treated with Bafilomycin A1. This effect was restored upon overexpression of Bnip3 in the knockout cells. Notably, when KCL22-NTC cells were subcutaneously injected into the flanks of mice, they gave rise to malignant tumors, unlike Ptbp2-KO-KCL22 cells. Also, transplantation of KCL22 cells through the tail vein in NOD/SCID mice resulted in higher cell engraftment and increased infiltration of malignant cells in the extramedullary organs. Our study underscores the role of PTBP2 in promoting cell proliferation and tumor formation while enhancing autophagy through Bnip3, thereby supporting the role of PTBP2 as an oncogene in CML. |
Publications
| Details |
|---|
List of publications A. During Ph.D
|
Group
| Details | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Alumnus/Alumna
Past M.Sc Project/Dissertation students, Project Technicians, and PDFs Dolly Hasda Dinesh Sahu Mumani Dash Nilima Dash Partha Sarathi Nathasharma Meera Kumari Ankita Mishra Pulak Mohanty Deeptimayee Padhy Sushanta Pradhan Manas Prusty Nayan Kumari Siba Prasad Mallick Puspendra Rout Lakesh Sahoo Suman Singh Amitesh Panda Ram Chandra Panigrahi Rahul Dey Ranjan Das Subhankar Behera Aditi Poddar Archana Ekka Ayushi Das Dr. Anurag Mitra Dr. Devi Sahoo Dr. Payal Guha Dr. Vivek Singh Dr. Yallamanda Vadlamudi Dr. Soloman Kabir Das, MBBS Dr. Soumika Sengupta
|














Grants
| Details | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Contacts
| Address | Fax | Office | |
|---|---|---|---|
| soumen@ils.res.in | Nalco Square, Bhubaneswar-751023, India | 0091 674 2300728 | 0674-2304327 |
Highlights
| Details |
|---|
Recently, we have applied for a patent. |
Positions
| Details |
|---|
|